La enfermedad de Kikuchi-Fujimoto (EKF), también conocida como linfadenitis necrosante no granulocítica o linfadenitis histiocítica necrosante, es una afección poco común pero significativa que merece una atención detallada. Descrita por primera vez en la literatura japonesa en 1972, esta enfermedad se caracteriza por ser una forma inusual de linfadenitis observada inicialmente en Japón, aunque su distribución geográfica se ha demostrado ser generalizada, con casos reportados en América, Europa y otras partes del mundo.
La etiología de la EKF sigue siendo desconocida, lo que añade un elemento de misterio a su naturaleza. Sin embargo, se han propuesto varias hipótesis, incluyendo factores infecciosos y autoinmunes. Algunos investigadores sugieren que la enfermedad podría ser el resultado de una reacción inmunológica del organismo desencadenada por infecciones virales, como el virus de Epstein-Barr (VEB), el Herpesvirus humano tipo 6, el Parvovirus B19 y el Citomegalovirus (CMV). Otras teorías apuntan a una respuesta inmune exagerada a diversos agentes o incluso a una predisposición genética, observándose con mayor frecuencia ciertos antígenos HLA de clase II en pacientes afectados.
La EKF afecta preferentemente a mujeres jóvenes, aunque puede presentarse en cualquier grupo de edad. Clínicamente, se manifiesta típicamente como una linfadenitis cervical dolorosa, a menudo acompañada de fiebre y leucopenia. Otros síntomas comunes incluyen mialgias, pérdida de peso, náuseas, vómitos, fatiga, sudación nocturna y exantemas. En ocasiones, puede presentarse con adenopatías generalizadas, afectando regiones como las axilares o supraclaviculares, e incluso, en casos raros, con afectación extranodal en órganos como riñones, hígado, tracto gastrointestinal, glándulas suprarrenales, médula ósea o el sistema nervioso central. La enfermedad suele tener un curso benigno y autolimitado, resolviéndose espontáneamente en un período de semanas a meses, aunque se han descrito recurrencias en un pequeño porcentaje de pacientes (aproximadamente 3-4%).
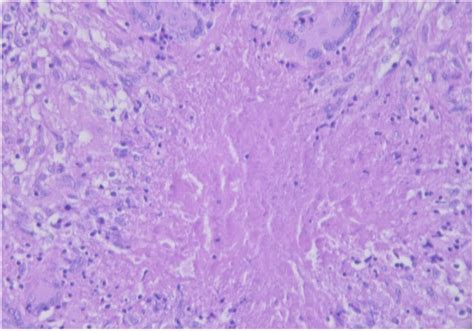
El diagnóstico definitivo de la EKF se basa en el examen histológico de las adenopatías. Las características histológicas clave incluyen fenómenos necróticos con cariorrexis, pérdida parcial de la estructura ganglionar y focos de histiocitos en la zona cortical y/o paracortical. Un hallazgo distintivo y crucial para diferenciarla de otras linfadenitis necrotizantes es la ausencia de granulocitos neutrófilos en los infiltrados inflamatorios. Se han descrito tres subtipos histológicos (necrotizante, proliferativo y xantomatoso), que se consideran reflejos de diferentes etapas evolutivas de la enfermedad.
El diagnóstico diferencial de la EKF es amplio y debe incluir otras afecciones como linfomas (incluyendo linfoma de células T/CD8+, que puede ser confundido erróneamente con EKF debido a la presencia de inmunoblastos y linfocitos atípicos), infecciones bacterianas (como tuberculosis), toxoplasmosis y lupus eritematoso sistémico (LES). La histología puede ser similar a la linfadenitis lúpica, y la presencia de cuerpos hematoxifílicos, depósito de ADN en la pared de los vasos o vasculitis fuera del área de necrosis pueden sugerir esta última.
La asociación entre la EKF y el lupus eritematoso sistémico (LES) es un aspecto importante a considerar. La EKF puede presentarse antes, después o simultáneamente al diagnóstico de LES. Aunque la etiología exacta de esta coexistencia es objeto de debate, algunos casos sugieren que la EKF podría ser una manifestación de la linfadenitis lúpica o un proceso concomitante. En estos casos, el seguimiento clínico y la vigilancia de posibles manifestaciones autoinmunes son fundamentales.
En cuanto al tratamiento, no existe una terapia específica para la EKF. Generalmente, la enfermedad se resuelve espontáneamente. En casos con síntomas severos, se puede recurrir al uso de antiinflamatorios no esteroideos (AINEs) o, en casos muy sintomáticos, esteroides a bajas dosis. La administración de antibióticos no es efectiva, ya que la enfermedad no es de origen bacteriano.
La presentación clínica de la EKF puede ser muy variada, lo que a veces dificulta el diagnóstico. Se han recogido casos de la enfermedad en diferentes localizaciones, como el mediastino anterior o la glándula parótida, y con presentaciones clínicas que van desde cuadros similares a enfermedades infecciosas hasta simulaciones de procesos neoplásicos. La enfermedad de Kikuchi-Fujimoto es una entidad que, a pesar de su rareza, debe ser considerada en el diagnóstico diferencial de linfadenopatías y fiebre, especialmente en mujeres jóvenes, dada su naturaleza benigna y la importancia de descartar patologías más graves.
Pacientes con patologías raras se ‘enferman’ con los trámites
Se han documentado casos de EKF en pacientes pediátricos, como una niña de 8 años con adenopatía cervical dolorosa, y un niño de 12 años con fiebre y tumoración cervical, demostrando la afectación en diferentes grupos de edad. La evolución en estos casos fue favorable, subrayando el curso generalmente benigno de la enfermedad.
En resumen, la enfermedad de Kikuchi-Fujimoto es una linfadenitis necrosante histiocítica, de etiología desconocida, que afecta predominantemente a mujeres jóvenes. Se caracteriza por linfadenopatías, fiebre y leucopenia, con hallazgos histopatológicos específicos. Su diagnóstico diferencial incluye diversas patologías, y su asociación con enfermedades autoinmunes como el LES es relevante. Aunque benigna y autolimitada, su correcta identificación es crucial para el manejo adecuado del paciente.
